Кетонурија е присуство во урината на кетонски тела, имено ацетон
Механизмот на кетонурија.
Кај здрава личност, јаглехидратите, липидите и некои протеини се оксидираат до јаглерод диоксид и вода. Во некои патолошки состојби, особено со дијабетес, производството на инсулин се намалува. Во црниот дроб, резервите на гликоген се намалени, многу телесни ткива се во состојба на глад на енергија. Под овие услови се активираат процесите на оксидација на протеините и липидите во црниот дроб, но недостатокот на гликоген доведува до нивно нецелосно оксидација и акумулација во крвта на неоксидираните производи на липидниот и протеинскиот метаболизам - тела на кетонски тела. Поради акумулацијата на кетонските тела во крвта (кетонемија), pH на крвта се префрла на киселинската страна. Оваа состојба се нарекува ацидоза. Урината на таков пациент има остра реакција на киселина и мириса на ацетон.
Глад, употреба на главно протеини и масна храна, исклучување на јаглени хидрати од диетата доведува до зголемено формирање на кетонски тела и нивно излачување во урината.
Во рана возраст, кетонурија е почеста отколку кај возрасните и нема клиничко значење. Овој феномен го интересира педијатарот со "ацетонемично повраќање" поврзано со грешки во исхраната.
Кетонурија во постоперативниот период се должи на распаѓање на протеините како резултат на хируршка траума.
Кај возрасните, кетонуријата се јавува во тешки форми на дијабетес и е со голема дијагностичка вредност. Кај децата, може да биде со разни болести, како резултат на лабилноста на метаболизмот на јаглени хидрати. Затоа, дури и мали грешки во исхраната, особено во присуство на акутна инфекција, нервозна возбуда, прекумерна работа, итн. може да доведе до кетоза. Кетонурија во раното детство може да се забележи со токсикоза, продолжено гастроинтестинално нарушување, дизентерија и други болести. Кај новороденчиња, зголемувањето на кетоните во урина скоро секогаш е предизвикано од неухранетост. Кетонурија, забележана кај заразни болести - шарлах треска, грип, туберкулозен менингитис и интоксикација, е секундарен знак, минлива и нема голема дијагностичка вредност.
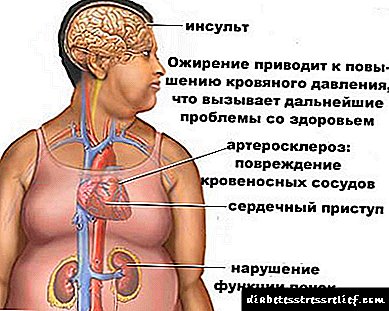
Кетонурија за дијабетес се развива заради зголемена кетогенеза и нарушена кетолиза. Зголемената кетогенеза доведува до зголемена мобилизација на маснотиите од масното ткиво, намалување на формирањето на оксалацетат во циклусот Кребс и намалување на биосинтезата на масни киселини. Во сериозен дијабетес, придружено со оштетување на ткивото на бубрезите (место за распаѓање на кетони), се јавува дополнително прекршување на кетолизата.
Отсуството на глукозорија во присуство на кетонурија го исклучува дијабетесот.
Нормално, кетонските тела се формираат во мала количина од крајниот производ на метаболизмот на јаглени хидрати и липиди - ацетил-CoA (C2-тела) преку ацетоцетил-CoA и се скоро целосно неутрализирани.
Кај дијабетес мелитус, мобилизацијата на липидите се зголемува со формирање на голема количина ацетил CoA (C2тело). во исто време, поради повреда на метаболизмот на јаглени хидрати, намалување на формирањето на оксалацетат, со кое2- телата се вклучени во циклусот Кребс и изгорени до јаглерод диоксид и вода. Покрај тоа, како резултат на зголемено разложување на липидите, обратната биосинтеза на Ц е блокирана.2тело во масни киселини. Така, се акумулира голема количина на Ц2-кој, што доведува до производство на голема количина на CoA, и следствено на тоа, ацетон, ацетоацетична и бета-хидроксибутирична киселина, кои се излачуваат во урината.
Тела на кетон
Овие се производи со средно распаѓање кои се формираат за време на функцијата на црниот дроб. Обично, метаболизмот ги изложува кетонските тела на понатамошна деградација.
Резервите на глукоза на телото се наоѓаат во маснотии во телото, поради што кетонуријата е силен недостаток на јаглени хидрати. Телата на кетон се одличен давател на енергија. Во ова, дури и масните киселини заостануваат зад нив. Затоа, кога на мозокот или срцето му недостасуваат фосфорни киселини, телото веднаш произведува кетонски тела со забрзано темпо.
Кои се причините
Кетонурија е содржина на урина во кетонски тела над нормалата.
Во здраво човечко тело, метаболизмот е добро избалансиран. Што може да предизвика кетонурија?

- Доминантноста на протеините и мастите во исхраната. Поради намалување на јаглехидратите во храната, клетките немаат исхрана. Наспроти ова, се развива кетонурија. Ова е реакцијата на телото на нерамнотежа во исхраната.
- Диетите и злоупотребата на глад можат да предизвикаат појава на кетонски тела во урината. Грешките во изборот на диети доведуваат до брзо разложување на маснотиите, додека бројот на ензими се зголемува. Кетонурија е исто така појава на ацетон во крвта. Обично, ако некое лице гладува повеќе од шест дена, содржината на кетонските тела во човечкото тело се зголемува над нормалното.
- Анестезија за операција.
- Дијабетес мелитус. Во овој случај, кетонурија се појавува од време на време. Телата кетонски сами по себе не се причина за заболувањето. Кај дијабетес мелитус, се пропишува дополнителен тек на јаглени хидрати и инсулин.
- Дехидрирање. Се јавува кога телото се прегрева или е компликација на дијабетес.
- Заболување на црниот дроб или сериозни заразни заболувања (на пр. Дизентерија).
- Развој на тумори во дигестивниот тракт.
- Нарушувања во панкреасот.
- Труење со алкохол или со такви хемиски соединенија како фосфор, олово.
- Оштетување на централниот нервен систем, нервна возбуда.
Запомнете дека мирисот на ацетон кога дишете или уринирате е сигнал за да видите лекар. Исто така, тоа е причина да се промени начинот на живот и исхраната.
Внимание на децата
Клиниката често се консултира доколку детето има повраќање, мирис на ацетон. Или таков мирис се појави во урината. И, иако е знак на кетонурија, тој не е нужно симптом на сериозна болест.

Честопати, сè е објаснето со несовршен метаболички систем. Причината за кетонурија кај деца е состојба во која се троши значителна количина на енергија. Обично се јавува кога:
- емоционална пресврт
- зголемен физички напор,
- неурамнотежена диета
- студ
Факт е дека телото на детето нема големи продавници на гликоген, затоа, се појавува активно разложување на маснотиите и се забележуваат знаци на кетонурија.
Бременост
За девет месеци од бременоста, жените честопати треба да полагаат тестови на урина. Ова е неопходно за да не пропуштите најмала девијација од нормата во телото за целиот период на гестација. Навистина, сите органи имаат голем товар. Што значи дека кетонските тела се наоѓаат во урината?
Обично нивната мала содржина е норма. Наједноставната анализа ќе го помине брзиот тест. Треба да ја спуштите лентата за тестирање во урината.
Негативниот тест се смета за нормален за време на бременоста или бројот на кетони е минимален. Ако вредноста на тестот е од 15 до 160 mg / dl - тоа е причина за загриженост.
За време на бременоста, кетонуријата е алармантен симптом. Се појавува со рана токсикоза. Труењето на телото, а со тоа и фетусот со ацетон, го комплицира текот на бременоста.

Ако бремената жена има сериозна хормонална слабост, таа е во опасност.
Зголемувањето на нивото на кетоните во урината на новороденче се должи на недоволно хранење или грешки во исхраната.
Симптоми на Кетонурија
Ако нивото на кетоните во телото значително се зголеми, тогаш кетонурија ќе се манифестира:
- исцрпувачка поспаност,
- мирис на ацетон од устата,
- мирис на урина ацетон
- анализата ќе покаже високо ниво на бели крвни клетки во крвта,
- биохемискиот тест на крвта ќе покаже мала содржина на гликоза,
Ако кетонурија предизвика скок во крвта на ацетон, тогаш може да се појави ацетон криза.
Значително зголемување на киселоста во клетките може сериозно да ги оштети внатрешните органи. Во овој случај, се започнува заштитна реакција - повраќање.
Симптоми на кетонурија кај деца:
- поплаки за болка во стомакот
- поплаки за главоболка
- детето е уморно, летаргично,
- поплаки за гадење
- повраќање
- покачување на температурата на 39 ° C,
- одбивање на храна
- мириса на ацетон
- зголемен црн дроб
Може ли да се открие кетонурија?
Само хемиската анализа може да открие зголемување на нивото на кетонските тела во организмот. Лабораторијата веднаш ќе ја утврди нормата на кетонските тела содржани во неа.
Во современата медицина, кетонурија е откриена:
- Тест на Ланге,
- Правен тест
- Пример Лестрада,
- модифициран примерок на Ротера,
- брзи тестови

Брзите тестови, се разбира, се најчести. Нивното дејство се заснова на хемиска реакција, чиј резултат е видлив скоро веднаш. Треба само да ја поставите лентата за испитување во урината или да ја испуштите на тест таблета. Во случај на позитивна реакција, тестовите стануваат виолетови. Осветленоста на бојата ви овозможува да процените според посебна скала во боја за тоа колку е надмината нормата на кетонските тела.
Меѓународна класификација на болести
Меѓународната статистичка класификација на болести и сродни здравствени проблеми (МКБ) е референтен водич преку кој материјалите можат да се споредат на меѓународно ниво. Со негова помош се комбинираат различни методолошки пристапи. Статистиката на болести се акумулира и класифицира. На секои десет години, IBC го разгледува комисијата на Светската здравствена организација. Со цел да се класифицира и анализира целата маса на акумулираните резултати од гледна точка на статистиката, таа се преведува на алфанумерички кодови. При таква работа се користи развиениот МКБ. Денес тоа одговара на МКБ-10. Содржи 22 паралелки (делови).
Според директориумот на МКБ, кетонурија има код R82.4.
Превенција и диета на кетонурија
За спречување на кетонурија, потребно е:
- јадат право
- водат здрав начин на живот:
- колку што е можно да се биде на свеж воздух,
- умерено вежбање
- Не започнувајте хронични заболувања.

Во хронично заболување како што е дијабетес, лекар се консултира систематски. Од време на време потребно е да се земе експресен тест.
Позитивна реакција укажува на патологија во телото. Обрнете внимание на сигналот! Итна жалба на клиниката брзо ќе го врати телото во нормала. Без да заминете од диетата пропишана од лекарот, можете да го забрзате закрепнувањето.
Принципи на диета на кетонурија:
- не јадете храна што е богата со протеини и маснотии,
- редовно внесување јаглени хидрати
- пијте повеќе раствори вода и сода (со дијабетес - инсулин).
Менито треба да содржи: варено говедско месо и зајак, најразлични супи од зеленчук, риба со малку маснотии. Корисни житарки без масло, зеленчук и овошје. Се препорачува да пиете повеќе сокови, овошни пијалоци, компоти.
Исклучи од менито:
- масно месо
- зачинети зачини
- слатко
- агруми
- банани
- печурки
- брза храна.
Фокус на третманот
Кетонурија не смее да се третира како посебна болест. Таа е само последица. Неопходно е да се исклучат причините што го предизвикале. Прво, потребно е целосно испитување. Точноста на дијагнозата и утврдувањето на причината за кетонурија гарантира успешен третман.
Лекарите даваат неколку совети:
- Ако имате прекумерна телесна тежина, треба да организирате денови за постот од време на време.
- Ако анализата открила зголемување на кетоните во урина, купете тестови за да ги користите дома.

- Урината треба да се собира за анализа не повеќе од четири часа пред породувањето.
- Детето може да се пие со алкален пијалок на секои 10-15 минути во мали делови. Активиран јаглен, ентеросел ќе помогне да се исчисти дигестивниот тракт.
- Со претстојно повраќање, корисно е да се пие фракционо пијалок. Повикајте брза помош.
Како да се третираат кетонурија, постојат совети во народната медицина.
- Со чести повраќање, пијте минерална вода, компот од суво овошје, раствор на гликоза. Една лажица за десет минути.
- Ставете клизма дома. Прво, вода на собна температура, подоцна топла, во која се додава лажичка сода.
- Пијте пијалок: растворете 2 лажици во 1 литар вода. мед, истурете го сокот од еден лимон. Пијте 1 лажица масло. на секои 15 минути.
- Рецепт за раствор од сода: растворете 1 лажичка сода во 250 ml вода. Пијте 1 лажиче. на секои 10 минути.
- Земете лушпи од смирувачки билки.
- За да се забрза елиминацијата на токсините од телото, јадете малку по малку. Подобро е да јадете само крекери.
Третманот зависи од индивидуалните карактеристики на телото. Мора да биде строго контролиран од присутните лекар.
76. холестерол. Начини на влез, употреба и екскреција од телото. Холестерол во серумот. Биосинтеза на холестерол, нејзините фази. Регулирање на синтезата.
Холестеролот е стероид специфичен за животинските организми. Се синтетизира во многу човечки ткива, но главното место на синтезата е црниот дроб. Во црниот дроб се синтетизира повеќе од 50% холестерол, во тенкото црево - 15-20%, остатокот од холестерол се синтетизира во кожата, кората на надбубрежната жлезда и гонадите. Околу 1 g холестерол се синтетизира на ден во организмот, 300-500 мг се ингестираат со храна (Сл. 8-65). Холестеролот извршува многу функции: тој е дел од сите клеточни мембрани и влијае на нивните својства, служи како почетен супстрат во синтезата на жолчните киселини и стероидните хормони. Прекусори во метаболичкиот пат на синтезата на холестерол, исто така, се претвораат во убикинон, компонента на респираторниот ланец и доликол, кој е вклучен во синтезата на гликопротеините. Поради својата хидроксилна група, холестеролот може да формира естри со масни киселини. Етерифициран холестерол преовладува во крвта и се чува во мали количини кај некои типови клетки кои го користат како подлога за синтеза на други супстанции. Холестеролот и неговите естри се хидрофобни молекули, така што тие се транспортираат преку крв само како дел од различни видови на лекови. Размената на холестерол е исклучително комплексна - само за нејзина синтеза, потребни се околу 100 последователни реакции. Вкупно, околу 300 различни протеини се вклучени во метаболизмот на холестерол. Нарушувања на метаболизмот на холестерол доведуваат до една од најчестите заболувања - атеросклероза. Смртноста од ефектите на атеросклероза (миокарден инфаркт, мозочен удар) води во целокупната структура на смртност. Атеросклерозата е „полигено заболување“, т.е. многу фактори се вклучени во неговиот развој, од кои најважните се наследните. Акумулацијата на холестерол во организмот доведува до развој на друга честа болест - болест на жолчни камења.
А. Синтеза на холестерол и негово регулирање
Реакциите на синтезата на холестерол се јавуваат во цитозолот на клетките. Ова е една од најдолгите метаболички патеки во човечкото тело.
Фенилкетонурија

Фенилкетонурија - наследно кршење на метаболизмот на аминокиселини поради недостаток на ензими на црниот дроб вклучени во метаболизмот на фенилаланин до тирозин. Раните знаци на фенилкетонурија се повраќање, летаргија или хиперактивност, мирис на мувла од урина и кожа, одложен психомоторен развој, типични доцни знаци вклучуваат олигофренија, доцнење на физички развој, конвулзии, егзематозни промени на кожата, итн. последователните дијагностика вклучуваат молекуларно генетско тестирање, определување на концентрација на фенилаланин во крвта, биохемиска анализа на урина, ЕЕГ и МРИ на мозокот. Третманот на фенилкетонурија треба да следи посебна диета.

Општи информации
Фенилкетонурија (Felling's болест, фенилпировична олигофренија) е конгенитална, генетски утврдена патологија која се карактеризира со нарушена хидроксилација на фенилаланин, акумулација на аминокиселини и неговите метаболити во физиолошки течности и ткива, проследена со сериозно оштетување на централниот нервен систем. Фенилкетонурија за прв пат е опишана од А. Сеча во 1934 година; се јавува со фреквенција од 1 случај на 10,000 новороденчиња. Во неонаталниот период, фенилкетонурија нема клинички манифестации, сепак, внесот на фенилаланин со храна предизвикува манифестација на болеста веќе во првата половина на животот, а последователно доведува до сериозни нарушувања во развојот на детето. Затоа пред-симптоматското откривање на фенилкетонурија кај новороденчиња е најважната задача на неонатологија, педијатрија и генетика.
Причини за фенилкетонурија
Фенилкетонурија е автозомно рецесивно нарушување на наследство. Ова значи дека за развој на клинички знаци на фенилкетонурија, детето мора да наследи една неисправна копија на генот од двајцата родители, кои се хетерозиготни носители на мутантниот ген.
Најчесто, развојот на фенилкетонурија е предизвикан од мутација во генот што го кодира ензимот фенилаланин-4-хидроксилаза сместен на долгата рака на хромозомот 12 (локус 12q22-q24.1). Ова е таканаречена класична фенилкетонурија од тип I, која претставува 98% од сите случаи на заболување. Хиперфенилаланинемија може да достигне 30 мг% и повисоко. Ако не се лекува, оваа варијанта на фенилкетонурија е придружена со длабока ментална ретардација.
Покрај класичната форма, се разликува атипична фенилкетонурија, продолжувајќи ги истите клинички симптоми, но не и подложен на корекција со диетална терапија. Тука спаѓаат фенилкетонурија тип II (недостаток на дехидодертерин редуктаза), фенилкетонурија тип III (недостаток на тетрахидробиоперин) и други, поретки варијанти.
Веројатноста за раѓање дете со фенилкетонурија се зголемува со блиски бракови.
Патогенеза на фенилкетонурија
Класичната форма на фенилкетонурија се заснова на инсуфициенција на ензимот фенилаланин-4-хидроксилаза вклучена во конверзијата на фенилаланин во тирозин во митохондрите на хепатоцитите. За возврат, дериватниот тирозин тирамин е почетен материјал за синтеза на катехоламини (адреналин и норепинефрин) и диодиодрозин за формирање на тироксин. Покрај тоа, формирањето на пиламентот на меланин е резултат на метаболизмот на фенилаланин.
Наследниот недостаток на фенилалаин-4-хидроксилаза ензим кај фенилкетонурија доведува до повреда на оксидација на фенилаланин од храна, што резултира во неговата концентрација во крвта (фенилаланинемија) и цереброспиналната течност значително се зголемува, а нивото на тирозин соодветно се намалува. Вишокот на фенилаланин се елиминира со зголемено уринарно излачување на неговите метаболити - фенилпировична киселина, фенилмилактична и фенилацетна киселина.
Нарушувањето на метаболизмот на аминокиселините е придружено со нарушено миелинирање на нервните влакна, намалување на формирање на невротрансмитери (допамин, серотонин, итн.), Активирање на патогенетски механизми на ментална ретардација и прогресивна деменција.
Симптоми на фенилкетонурија
Новороденчињата со фенилкетонурија немаат клинички знаци на болеста. Обично, манифестацијата на фенилкетонурија кај деца се јавува на возраст од 2-6 месеци. Со почетокот на хранењето, протеинот на мајчиното млеко или неговите заменици почнува да влегува во телото на бебето, што доведува до развој на првите, неспецифични симптоми - летаргија, понекогаш вознемиреност и хипер ексцитабилност, регургитација, мускулна дистонија, конвулзивен синдром. Еден од раните патогномонични знаци на фенилкетонурија е постојаното повраќање, кое често погрешно се смета за манифестација на пилорна стеноза.
До втората половина од годината, застојот на детето во психомоторниот развој станува забележлив. Детето станува помалку активно, рамнодушно, престанува да ги препознава најблиските, не се обидува да седне и да застане на нозе. Абнормалниот состав на урина и пот предизвикува карактеристичен мирис на глувчето (мирис на мувла) што потекнува од телото. Често се појавува пилинг на кожата, дерматитис, егзема, склеродермија.
Кај деца со фенилкетонурија кои не примаат третман, се откриени микроцефалија, прогнотија, подоцна (по 1,5 години) почетнички, се открива емајл хипоплазија. Забележано е одложување на развојот на говорот, а за 3-4 години се открива длабока олигофренија (идиотизам) и речиси целосно отсуство на говор.
Децата со фенилкетонурија имаат диспластична телесна форма, често вродени срцеви мани, автономни дисфункции (потење, акроцијаноза, артериска хипотензија) и страдаат од запек. Фенотипските карактеристики на децата кои страдаат од фенилкетонурија вклучуваат лесна кожа, очи и коса. Дете со фенилкетонурија се карактеризира со специфична поза на „кројач“ (горните и долните екстремитети свиткани во зглобовите), тремор на рацете, волнено движење, мелено одење и хиперкинеза.
Клиничките манифестации на фенилкетонурија од типот II се карактеризираат со сериозен степен на ментална ретардација, зголемена раздразливост, напади, спастична тетрапареза и хиперрефлексија на тетива. Прогресијата на болеста може да доведе до смрт на дете на возраст од 2 до 3 години.
Кога фенилкетонури тип III развива тријада на знаци: микроцефалија, олигофренија, спастична тетрапареза.
Дијагноза на фенилкетонурија
Во моментов, дијагнозата на фенилкетонурија (како и галактоземија, вроден хипотиреоидизам, адреногенитален синдром и цистична фиброза) е дел од програмата за неонатална скрининг за сите новороденчиња.
Скрининг-тест се спроведува на 3-5 дена од животот на целосен рок и 7 дена од животот на предвремено бебе со земање примерок од капиларна крв на посебна форма на хартија. Доколку се открие хиперфенилаланемија, повеќе од 2,2 мг% од детето се упатува на детска генетика за преиспитување.
За да се потврди дијагнозата на фенилкетонурија, се проверува концентрацијата на фенилаланин и тирозин во крвта, се утврдува активност на хепатални ензими (фенилаланин хидроксилаза), се врши биохемиско испитување на урината (определување на кетонски киселини), метаболити на катехоламини во урината и слично, се изведува активност на хепатални ензими (фенилаланин хидроксилаза), биохемиски преглед на урина (определување на кетонски киселини), метаболити на катехоламини во урината, итн.
Генетски дефект на фенилкетонурија може да се открие дури и за време на бременоста за време на инвазивна пренатална дијагноза на фетусот (хорионбиопсија, амниоцентеза, кордоцентеза).
Диференцијалната дијагноза на фенилкетонурија се спроведува со интракранијална траума при раѓање кај новороденчиња, интраутерини инфекции и други метаболички нарушувања на аминокиселини.
Третман на фенилкетонурија
Основен фактор во третманот на фенилкетонурија е диета што го ограничува внесот на протеини во организмот. Третманот се препорачува да започне со концентрација на фенилаланин> 6 мг%. Развиени се специјални мешавини за новороденчиња - Афенилак, Лофенилак, за деца над 1 година - Тетрафен, без фенил, над 8 години - Максимум-XP и други.Основа на диетата е храна со малку протеини - овошје, зеленчук, сокови, протеински хидролизати и аминокиселини мешавини . Проширувањето на диетата е можно по 18 години во врска со зголемување на толеранцијата кон фенилаланин. Во согласност со рускиот закон, обезбедувањето медицинска исхрана за луѓето кои страдаат од фенилкетонурија треба да биде бесплатно.
Пациентите се пропишани внес на минерални соединенија, витамини од групата Б, итн., Според индикациите - ноотропни лекови, антиконвулзиви. Во сложената терапија на фенилкетонурија широко се користат општа масажа, вежбање и акупунктура.
Децата кои страдаат од фенилкетонурија се под надзор на локален педијатар и невропсихијатар, и честопати им е потребна помош од логопед и патолог. Потребно е внимателно следење на невропсихичкиот статус на децата, следење на нивото на фенилаланин во крвта и индикатори на електроенцефалограм.
Атипични форми на фенилкетонурија кои не се подложни на третман со диети, бараат назначување на хепатопротектори, антиконвулзиви, терапија со замена со леводопа, 5-хидрокситрипптофан.
Предвидување и спречување на фенилкетонурија
Спроведување на масовен скрининг за фенилкетонурија во новороденото време ви овозможува да организирате рана диетална терапија и да спречите сериозно церебрално оштетување, нарушена функција на црниот дроб. Со раното назначување на елиминационата диета за класична фенилкетонурија, прогнозата за развој на деца е добра. Со доцна третман, прогнозата за ментален развој е лоша.
Превенција на компликации на фенилкетонурија се состои во масовно скринирање на новороденчиња, рано препишување и долгорочно усогласување со диеталната исхрана.
За да се процени ризикот од раѓање дете со фенилкетонурија, прелиминарно генетско советување треба да им се даде на парови кои веќе имаат болно дете, се во конгениозна врска и имаат роднини со оваа болест. Womenените со фенилкетонурија кои планираат бременост треба да следат строга диета пред зачнувањето и за време на бременоста за да се исклучи зголемувањето на нивото на фенилаланин и неговите метаболити и нарушен развој на генетски здрав фетус. Ризикот да имате дете со фенилкетонурија кај родители кои носат неисправен ген е 1: 4.





-
Методи за превенција на дијабетес
Методи за превенција на дијабетес Според СЗО, бројот на луѓе кои страдаат од дијабетес е близу 300 милиони.Ова е околу 6% од популацијата, во која спаѓаат возрасните категории од 20-79 години. ... -

-

-