Терапевтска архива бр 03 2018 - Хемохроматоза - тековната состојба на проблемот

Хемохроматозата е наследна патологија поврзана со голема апсорпција на железо во органите за варење и нејзино последователно прекумерно акумулирање во разни внатрешни органи.
Црниот дроб страда повеќе од другите. Раното откривање на хемохроматоза, нејзината дијагноза и третман нема да дозволи развој на последици.
Хемохроматоза - модерна состојба на проблемот
Н.Б. ВОЛОШИНА1, М.Ф. ОСИПЕНКО1, Н.В. ЛИТВИНОВА 1, А.Н.ВОЛОШИН2
1 Државен медицински универзитет во Новосибирск ФГБУ во НСМУ на Министерството за здравство на Русија, Русија,
Градска клиничка болница 2 Новосибирск 2, Русија
Синдром на преоптоварување со железо може да биде поврзан со разни стекнати состојби и наследни фактори. Наследната хемохроматоза е најчестото генетско нарушување. Без терапевтска интервенција болеста може да доведе до развој на опасни по живот компликации како цироза, хепатоцелуларен карцином. Написот презентира податоци за патогенезата, дијагнозата и третманот на наследна хемохроматоза. Дадено е сопствено клиничко набудување.
Клучни зборови: наследна хемохроматоза, третман, флеботомија.
 Хемохроматозата е болест поврзана со акумулацијата на високи патолошки нивоа на железо во организмот, што доведува до функционални нарушувања на некои органи. Обично, апсорпцијата на железо е цврсто регулирана, како резултат на што телото не е во можност да лачи вишок железо. Вишокот на железо се акумулира во клетките како hemosiderin. Ова на крајот доведува до смрт на клетките и замена на овие клетки со фиброзно ткиво, што доведува до нарушување на структурата и функцијата на органите. Со хемохроматоза, можно е оштетување на црниот дроб, панкреасот, срцето, тироидната жлезда, зглобовите, кожата, гонадите и хипофизата.
Хемохроматозата е болест поврзана со акумулацијата на високи патолошки нивоа на железо во организмот, што доведува до функционални нарушувања на некои органи. Обично, апсорпцијата на железо е цврсто регулирана, како резултат на што телото не е во можност да лачи вишок железо. Вишокот на железо се акумулира во клетките како hemosiderin. Ова на крајот доведува до смрт на клетките и замена на овие клетки со фиброзно ткиво, што доведува до нарушување на структурата и функцијата на органите. Со хемохроматоза, можно е оштетување на црниот дроб, панкреасот, срцето, тироидната жлезда, зглобовите, кожата, гонадите и хипофизата.
Преоптоварувањето на железо, што предизвикува хемохроматоза, може да се случи на три начина: масивно внесување на железо од железо, зголемена апсорпција на железо при нормален внес на железо и прекумерно производство или масовна, честа трансфузија на црвени крвни клетки.
Кај наследната хемохроматоза, вишокот железо обично се депонира во паренхимните клетки, додека при трансфузија хемохроматоза главно се депонира во ретикулоендотелијалните клетки 1-3.
Наследната хемохроматоза вклучува група на генетски нарушувања, кои се карактеризираат со зголемена апсорпција на железо. Доминантен механизам кај повеќето видови на наследна хемохроматоза е ефектот на хепцидин, кој игра клучна улога во железната хомеостаза 4-6. Хепсидин се синтетизира главно во хепатоцитите и ја контролира концентрацијата на железо во плазмата со врзување за феропортин (исто така наречен SLC40A1), единствениот познат трансмембранозен транспортер на железо од ткивата на донатори на железо. Феропортин извезува железо од дуоденумот, од макрофаги и хепатоцити.
Во плазмата, железото се врзува за трасферин, така што заситеноста на железо со трансферин е во просек 35% (просечна утринска вредност). Хепсидин спречува ослободување на железо од макрофаги (од стари црвени крвни клетки и феритин), хепатоцити и дуоденални ентероцити со врзување за феропортин. И во отсуство на феропортин, излезот на железо од ентероцити, хепатоцити и макрофаги е блокиран. Така, хепцидин ја намалува апсорпцијата на железо во цревата, го намалува нивото на ослободено железо од хепатоцитите и макрофагите, што доведува до ниско ниво на железо во плазмата и зголемување на ткивата.
Причината за наследна хемохроматоза е мутација во генот HFE. Дефектот во генот HFE за прв пат е опишан во 1996 година, што е мутација што доведува до замена на тирозин со цистеин во аминокиселинска позиција 282 (C282Y). Мутација во генот HFE предизвикува зголемена апсорпција на железо и покрај нормалното внесување железо. Протеинот HFE го регулира производството на хепатидин. Пациенти со наследна хемохроматоза хомозиготни C282Y се од 80 до 85% 1, 8.
Постојат уште две мутации: едната е поврзана со замена на аспартат со хистидин на позицијата 63 (H63D), а втората е замена на цистеин со серин на позиција 65 (S65C). Овие мутации не се поврзани со синдромот на преоптоварување со железо, освен ако C282Y е составен дел на хетерозиготите C282Y / H63D или C282Y / S65C. Така, форма на наследна хемохроматоза поврзана со HFE може да се потврди со асимптоматски тек на болеста. Соодветно на тоа, генетска дијагноза може да се примени кај пациенти кај кои хемохроматозата сè уште не се манифестирала фенотипски. Оваа група пациенти со генетска предиспозиција за хемохроматоза. Хетерозиготите имаат зголемен ризик од развој на дијабетес во споредба со општата популација, механизмот на развој е непознат 9-11.
Претходно се сметаше дека кај сите пациенти со дефект на генот HFE, со тек на време ќе се развие клиника за хемохроматоза. Како и да е, сега е откриено дека фенотипскиот израз се наоѓа само кај приближно 70% од хомозиготите на C282Y, а помалку од 10% од нив развиваат тежок преоптоварен железо со оштетување на внатрешните органи 12, 13.
Табелата ја покажува класификацијата на синдромите на преоптоварување со железо во зависност од причината за нејзиното појавување.
Во зависност од причината на болеста, пациентите со синдром на преоптоварување со железо можат да се поделат во 4 групи: пациенти со наследна хемохроматоза, пациенти со секундарна хемохроматоза предизвикана од разни причини и мала група на пациенти, што се издвојува како „различни“.
Причината за секундарна хемохроматоза е еритропоетска хемохроматоза. Најчесто ова се јавува како резултат на основно крвно заболување во кое црвените крвни клетки имаат пократок животен век. Оваа група на болести вклучува анемија на недостаток на железо, таласемија, сидеробластична анемија, хронична хемолитичка анемија, апластична анемија, чувствителна на пиридоксин анемија, дефицит на пироват киназа.
Синдром на преоптоварување со железо може да се појави кај пациенти кои примаат продолжено и повеќекратно трансфузија на црвени крвни клетки. Како што може да се види од табелата, други прилично ретки болести, како на пример, порфирија, исто така, можат да предизвикаат синдром на преоптоварување со железо.
Конечно, прекумерното внесување железо може да предизвика хемохроматоза. Добро познат историски факт: употребата на пиво направено во челични тапани беше причина за синдром на преоптоварување со железо. Исто така, предозирање со препарати од железо може да предизвикаат синдром на преоптоварување со железо. Мора да се запомни дека многу додатоци во исхраната без рецепт содржат железо во доволно голема доза, така што нивната неконтролирана употреба е неприфатлива.
Симптомите на болеста зависат од органот кој е најмногу зафатен, сепак, скоро сите пациенти се жалат на значителна слабост и замор. Нема специфични симптоми на хемохроматоза. Најчесто, дијагнозата се поставува во фаза на болеста, кога веќе се зафатени неколку системи. Од првите симптоми на болеста до верификација на дијагнозата обично трае најмалку десет години. Кај жени со хемохроматоза, симптомите на болеста се манифестираат во подоцнежна возраст отколку кај мажите, поради менструална загуба на крв, губење на „мајчино железо“ за време на бременоста и антиоксидантно дејство на естрогенот, а болеста клинички не се манифестира пред климактерискиот период.
Приближно 50% од пациентите со симптоми на наследна хемохроматоза имаат дијабетес мелитус, ризикот од нејзино појавување значително се зголемува кај хетерозиготните. Цироза на црниот дроб е присутна кај 70% од пациентите со хемохроматоза. Во оваа група на пациенти, инциденцата на хепатоцелуларен карцином, што е водечка причина за смрт, е значително зголемена.
Оштетување на зглобовите со хемохроматоза се манифестира во форма на артралгија (обично вториот и третиот метакарпофангеален зглоб). Деформитети на зглобовите со хемохроматоза обично не се случуваат, иако можни се дегенеративни зглобови. Кај овие пациенти, како по правило, кристалите на калциум пирофосфат може да се најдат во синовијалната течност. Карактеристично е за полиартритис со хемохроматоза што дури и по нормализирање на продавниците за железо, сепак може да напредува.
Депонирање на железо во влакна на срцевиот мускул и клетките на системот за спровод на срцето може да доведе до нарушување на срцевиот ритам и / или проширена кардиомиопатија, со понатамошен развој на срцева слабост. Во некои случаи, постои целосен компензација за откажување на левата комора по нормализирање на нивото на железо во телото 9-12.
Со хемохроматоза, можен е развој на хипогонадизам и, соодветно на тоа, импотенција поради хипоталамална и / или хипофизарна инсуфициенција, што доведува до повреда на ослободување на хормонот гонадотропин. Во случаи на вишок на железо продавници пет пати или повеќе, се јавува хиперпигментација на кожата, што е резултат на таложење на железо и меланин. Преоптоварувањето на железо на макрофагите може да доведе до нарушен фагоцитоза и намален имунитет, што доведува до зголемен ризик од инфекција од Листерија, Yersinia enterocolitica и Vibrio vulnificus. Депонирање железо во тироидната жлезда обично предизвикува хипотироидизам.
Развиената фаза на хемохроматоза се карактеризира со присуство на цироза, дијабетес мелитус и пигментација на кожата (т.н. бронзен дијабетес). Кај пациенти кои злоупотребуваат алкохол и се заразени со хепатитис Б и / или Ц, патологијата на црниот дроб и панкреасот поврзан со хемохроматоза се одвива значително потешко 1-3.
На дијаграмот се прикажани дијагностички мерки за сомнителна хемохроматоза. Познато е дека само околу 70% од хомозиготите на C282Y имаат покачено ниво на феритин, што одговара на зголемување на продавниците за железо, а само мал процент од овие пациенти имаат клинички манифестации на болеста. Се разбира, сите пациенти со симптоми кои можат да се појават со хемохроматоза, треба да поминат натамошно испитување за да се исклучи болеста. Особено внимание треба да се посвети на пациенти со немотивирана слабост, артралгија, болка во горниот десен квадрант на абдоменот, импотенција, намалено либидо, синдром на срцева слабост, пигментација на кожата и дијабетес. Покрај тоа, кај сите пациенти со хепатомегалија, цитолитичен синдром, со цирозната фаза на болеста, неопходно е, покрај сите можни етиолошки причини на болеста, да се запамети и можноста за хемохроматоза. Се разбира, наследната хемохроматоза треба да се исклучи кај пациенти со роднини од прв степен на сродство кои страдаат од хемохроматоза. 
Студијата треба да започне со мерење на заситеноста на серумската концентрација на трансферин или серумска феритин. Треба да се напомене дека определувањето на трансферин во случаи на еритропоетичка хемохроматоза не е толку ефикасно за верификација на синдром на преоптоварување со железо. Специфичноста на феритин во голема мерка зависи од присуството на воспалителни заболувања. Ако нивото на феритин е повисоко од 200 μg / l кај жени или 300 μg / l кај мажите или заситеноста на трансферин е повеќе од 40% кај жени или 50% кај мажите, потребно е дополнително тестирање за да се исклучи хемохроматоза 1, 2, 10, 11.
Според препораките на Американското здружение за проучување на заболувања на црниот дроб 2011 година (AASLD 2011) доколку пациентот има серум трансферин од 1000 мг / л), а во зависност од овие индикатори се донесува одлука за терапевтската тактика и потребата од биопсија на црниот дроб (види графикон )
Кај пациенти со комбинација на хетерозиготни C288Y / H63D, како и C288Y хетерозиготни или не C288Y, потребно е внимателно елиминација на други заболувања на црниот дроб или крв (доколку е потребно, неопходна е пункција за биопсија на црниот дроб), а потоа се донесува одлука за терапевтско крварење.
Не постојат веродостојни докази дека одредени диети влијаат на почетокот или прогресијата на хемохроматозата. Сепак, некои автори веруваат дека на пациентите со наследна хемохроматоза им е прикажана диета со исклучок на чај и агруми, кои, според нивното мислење, придонесуваат за акумулација на железо. Се разбира, алкохолот, кој е главната хепатотоксична супстанција, треба строго да се забрани за пациенти со хемохроматоза. Покрај тоа, етанол е докажано дека ја намалува синтезата на хепатин 20, 21.
Примарниот третман за примарна хемохроматоза е крварење. Намалување на бројот на црвени крвни зрнца, кои се главен мобилизатор на железо во организмот, со што се намалува и минимизира токсичниот ефект на железо. Пациентите може да бараат 50-100 крвни пробива годишно, по 500 ml секоја за да ги намалат нивоата на железо во нормала. Откако ќе се нормализира нивото на железо, потребно е доживотно, но поретко крварење, обично 3-4 пати годишно. Целта на крварењето е да се одржи нивото на феритин од 50-100 µg / L. Во случаи на значително намалување на хемоглобинот по крварење, препорачливо е третман на зглобовите со еритропоетин.
Ако се открие хемохроматоза во рана фаза на болеста, третманот со крварење може да спречи дисфункција на засегнатите органи и со тоа да го зголеми животниот век на пациентот. Сепак, пациентите ретко живеат повеќе од две години по дијагностицирањето, во случаи на доцна дијагностицирање во фаза на детални клинички манифестации 22, 23.
Според Европската асоцијација за проучување на црниот дроб (EASL 2010), индикациите за терапевтско крварење се покачени нивоа на серумски феритин. Се препорачува терапевтско крварење со волумен од 400-500 ml еднаш неделно или еднаш на 2 недели се додека не се достигне ниво на феритин од 45% и значително зголемување на серумскиот феритин до 1444 mcg / l, дијагнозата на хемохроматоза е непобитен. ДНК примероците беа анализирани за мутации во генот HFE - беше откриена мутација C282Y (c.845 G> A) во хомозиготна состојба s.845A / s.845 А.
Така, дијагнозата на пациентот К е наследна хемохроматоза, хомозиготна мутација во генот HFE (C288Y / C288Y) со доминантно оштетување на црниот дроб, фиброза од 1 степен (FibroScan, Метавир 6,6 kPa).
Доцната манифестација и дијагностицирање на болеста на 58-годишна возраст во 2015 година се должи на долгорочното компензација на болеста заради масовно губење на крвта поради менструална крв, донирање на крв и загуба на крв за време на прекинување на бременоста и породувањето.
Вреди да се одбележи дека поминаа 8 години од појавата на првите знаци на болеста до верификација на дијагнозата! Од крајот на 2015 година, на пациентот му е препишана терапија - крварење од 500 мл еднаш неделно. Пациентот добро го толерирал крварењето, забележал значително подобрување во состојбата по првата постапка. Беше следи општ тест на крвта и феритин во крвта, чие ниво постепено се намалуваше. Севкупно, повеќе од 100 крварења беа извршени за 2 години, сепак, до денес, целното ниво на трансферин (100 μg / l) не е постигнато, поради фактот што пациентот периодично ја прескокнува постапката, објаснувајќи ја нејзината здравствена состојба. Во моментов, пациентот продолжува со терапијата, таа успеа да ја убеди во потребата од доживотна терапија.
Така, мора да се запомни дека во присуство на цитолитичен синдром кај пациенти, наследната хемохроматоза треба да биде вклучена во дијагностичкиот претрес. Терапијата по избор на наследна хемохроматоза во моментов останува крварење. Соодветната терапија започната со тек на време овозможува да се избегне развој на цирозната фаза на болеста и со тоа да се зголеми животниот век на пациентите.
Информации за авторите:
Волошина Наталија Борисовна - кандидат за медицински науки, вонреден професор пропадеутика на внатрешни заболувања на медицинскиот факултет
Осипенко Марина Федоровна - доктор по медицински науки, проф. раководител. кафе пропадеутика на внатрешни заболувања на медицинскиот факултет
Волошин Андреј Николаевич - Доктор на градската клиничка болница во Новосибирск 2
Хемохроматоза: која е оваа болест?
За да ја разберете суштината на болеста, треба да знаете колку железо треба да има лицето нормално. Кај мажите, железото е околу 500-1500 мг, а кај жените, од 300 до 1000 мг. Индикаторите не зависат само од полот, туку и од тежината на личноста. Повеќе од половина од вкупната количина на железо е во хемоглобин.
Околу 20 мг од овој микроелемент влегува во телото со храна дневно. Од нив, само 1-1,5 мг се апсорбира во цревата. Со хемохроматоза (ГЦ) или сидерофилија, како што се нарекува и оваа болест, апсорпцијата се зголемува на 4 мг на ден, а железото постепено се акумулира во ткивата на разни органи.
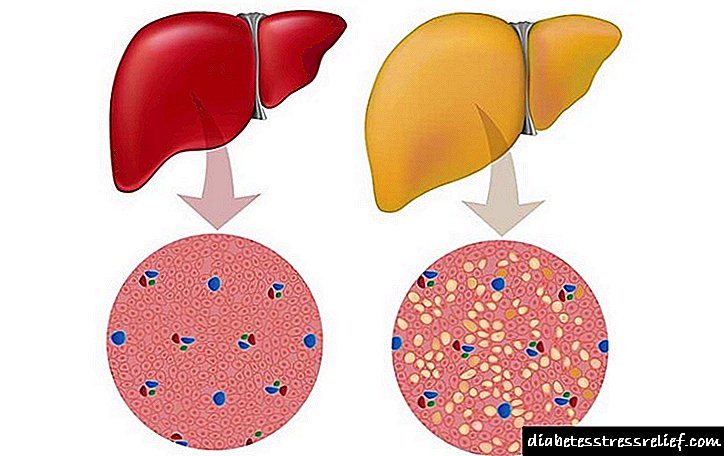
Здрав црн дроб и хемохроматоза
Неговиот вишок ги уништува молекулите на протеините и јаглехидратите, а со тоа и самиот орган. Кај пациенти со ГЦ, количината на железо во црниот дроб може да достигне 1% од сувата маса на органот, што е полн со цироза, а во третина од случаи со карцином на црниот дроб. Оштетено од вишок железо, панкреасот може да даде поттик за развој на дијабетес.
Депонирани во хипофизата, железото го уништува целиот ендокриниот систем. Репродуктивните органи страдаат повеќе од другите: мажите имаат еректилна дисфункција, а жените можат да развијат неплодност.
Причини за појава
Главната причина за ГЦ е „дефектно функционирање“ на генот, поточно, генот HFE. Тој е тој што го регулира текот на хемиските процеси и количината на железо што влегува во телото како дел од храна. Мутацијата што се јавува кај него доведува до нарушување на метаболизмот на железо.
Други причини за ГЦ се:
- таласемија. Во овој случај, структурата на хемоглобинот е уништена со ослободување на железо, т.е.
- хепатитис
- железо може да се зголеми како резултат на чести трансфузии на крв. Факт е дека животниот век на туѓите црвени крвни зрнца е многу пократок од нивниот. Кога умираат, ослободуваат железо,
- процедури на хемодијализа.
Код и класификација на МКБ-10
Во општо прифатениот класификатор на болести на ГЦ, е назначен код E83.1.
Во етилогична вена, се разликуваат примарни (или наследни ГК) и секундарни:
- примарно. Овој вид на болест има наследна природа и е резултат на дефект на ензимскиот систем кој влијае на метаболизмот на железо. Се дијагностицира кај 3 лица од 1000. Забележано е дека мажите се поподложни на оваа патологија и страдаат од тоа 3 пати почесто од жените,
- секундарно. Нејзината причина е заболувања на црниот дроб на пациентот (што често се забележува со алкохолизам), трансфузија на крв, само-третман со витамин комплекси со висока содржина на железо. Причината за стекната GC може да биде проблеми со кожата и болести на крвта.
Примарната хемохроматоза (PCH) се карактеризира со постепен развој, а во раните фази, пациентите се жалат на замор. Може да им пречи болка во десната страна и сува кожа.
Проширената фаза на PCH се карактеризира со:
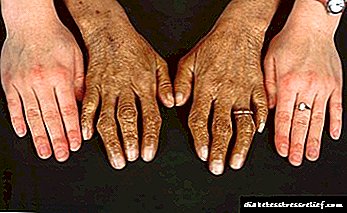
- специфична пигментација на лицето, вратот, рацете и пазувите. Тие заземаат бронзена нијанса,
- цироза на црниот дроб. Дијагностициран е во 95% од случаите,
- срцева слабост
- артритис
- дијабетес мелитус: во 50% од случаите,
- зголемена слезина,
- сексуална дисфункција.
Во последните фази се забележува портална хипертензија и асцити. Може да се развие карцином на црниот дроб.
 Бидејќи вишокот на железо се формира со текот на годините, почетните симптоми на секундарно GC се манифестираат кај мажи по 40 години, а кај жени по 60 години.
Бидејќи вишокот на железо се формира со текот на годините, почетните симптоми на секундарно GC се манифестираат кај мажи по 40 години, а кај жени по 60 години.
Симптомите се како што следува:
- мелазма,
- замор и слабеење,
- намалено либидо
- проширување и згуснување на ткивото на црниот дроб,
- цироза (во последната фаза на ГЦ).
Тест на крвта и други дијагностички методи
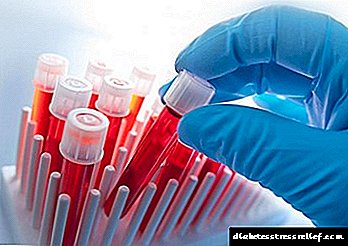 Гастроентеролог ја потврдува дијагнозата. Во раните фази на болеста, лабораториските тестови се многу важни.
Гастроентеролог ја потврдува дијагнозата. Во раните фази на болеста, лабораториските тестови се многу важни.
Со GC, се вршат посебни тестови на крвта за откривање на вредностите на железо во плазмата, неговата мала способност за врзување железо и заситеност со трансферин.
Главниот знак на заболувањето е наслаги на хемозидерин во хепатоцитите на црниот дроб, во кожата и другите органи, кои стануваат „рѓосани“ заради вишокот на овој пигмент. Општ тест на крвта е исто така потребен за биохемија, како и шеќер. Покрај тоа, се земаат тестови на црниот дроб.
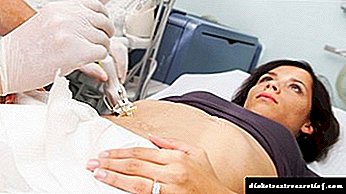 Дополнително, се вршат и инструментални студии:
Дополнително, се вршат и инструментални студии:
- биопсија на црниот дроб е главниот начин да се потврди ГЦ,
- Ултразвук на абдоменот
- МНР на црниот дроб (во некои случаи)
- ехокардиографија, за да се исклучи / потврди кардиомиопатија,
- радиографија на зглобовите.
Терапевтска диета
Важно е да се разбере дека со дијагностицирана хемохроматоза, диетата треба да биде доживотна.
Главното правило е максимално намалување на исхраната на производи што содржат железо, особено:

- тврди сирења и морска риба,
- житарки: овес, просо и леќата,
- црн леб
- мешунки и суво овошје,
- аскорбинска киселина и лекови со голема содржина на витамин Ц,
- Offal, особено црниот дроб, е целосно исклучен.
Алкохолот е апсолутен табу. Но, чајот и кафето, напротив, се прикажани. Тие имаат танин, што ја успорува апсорпцијата на железо.
Список на употребени лекови
Овој третман се спроведува со лекови кои отстрануваат железо од телото на пациентот. Во почетната фаза, се препишуваат витамини А, Е и фолна киселина. Потоа се користат хелатори (како што е Десферал).

Дозирање на инјектирање: 1g / ден. Веќе 500 мг од лекот дава опиплив резултат: се излачува до 43 мг железо. Курсот трае до 1,5 месеци. Продолжената употреба е опасна: заоблување на леќи е можно.
Флеботомија и други терапевтски процедури
 Флеботомијата е наједноставниот и, во исто време, доста ефикасен не-фармаколошки третман на ГК.
Флеботомијата е наједноставниот и, во исто време, доста ефикасен не-фармаколошки третман на ГК.
Се прави пункција во вената на пациентот и се ослободува крв од телото. Се исцедат околу 500 мл неделно.
Постапката е само амбуланта. Крвта постојано се тестира за концентрација на ферин: треба да се спушти до 50. Ова може да трае 2-3 години. Понатаму, терапијата е насочена кон одржување на оптимална вредност на овој елемент во трагови.
Третман со народни лекови
Оваа терапија има благ ефект врз заболените органи.
Третман на црниот дроб:

- тиква. Добро е и сурово и печено. Зеленчукот се додава во салатите или се меша со мед - вкусен и здрав! Прикажан е и сок од тиква: половина чаша на празен стомак,
- цвекло- Друг корисен производ за ГК. Користете во сурова или варена форма. Здрав и свежо исцеден сок.
За третман на срце, можете да советувате инфузии од глог, адонис или матичница. Билките се истураат со врела вода и, по инсистирање, се пијат според упатствата.
Третман на панкреас:

- лушпа од хлебните семе ќе помогне. Пропорции: 1 суп.л-патки. суровини до 1 суп.л-патки. вода. Сварено семе се вари 5 минути, се лади и се зема пред јадење, 1 лажица масло.,
- мед со цимет. Пропорции: 1 суп.л-патки. прав до 1 лажица вода. Инсистирајте 15-30 минути. и додадете малку мед. Оставете уште 2 часа. Сите средства треба да се пијат за еден ден.
Корисна и неварена овесна каша (со лушпа). Пропорции: 100 гр житни култури до 1,5 литри вода. Се вари најмалку половина час. Потоа, право во чинијата каде што се готви овесот, смачкајте го додека не се исцрпи и повторно ќе зоврие 40 минути. Theивотот на филтрираната супа не е повеќе од 2 дена. Пијте половина чаша пред јадење.
Прогноза и главни клинички упатства
Но, ако терапијата се спроведува под медицински надзор и на време, тогаш животот на пациентот значително се зголемува.
Како наследна болест, хемохроматозата во 25% од случаите се дијагностицира кај роднините на пациентот. Значи, тие мора да бидат испитани понатаму. Ова ќе ја открие болеста дури и пред клинички манифестации и во иднина да се избегнат нејзините компликации.
Во случај на секундарна GC, се препорачува диета, важно е да се задржи состојбата на црниот дроб и крвта под контрола. Хемохроматозата откриена за време на бременоста (или во фаза на планирање) не е опасна.
Поврзани видеа
За симптомите, причините и методите на лекување на хемохроматоза во видеото:
За жал, основната причина за хемохроматоза сè уште не е идентификувана. Но, во моментов е развиена и активна применета посебна сеопфатна техника на третман, чија цел е да се прекинат клиничките манифестации на болеста и да се намали ризикот од неговите можни компликации.
- Стабилизира нивото на шеќер подолго време
- Враќа производство на панкреас инсулин
Дознајте повеќе. Не е дрога. ->
Истовремена терапија на болести
Прекумерното железо во органите доведува до развој на повеќе патологии. Сите од нив бараат дополнителна терапија. На пример, ако ГК придонесе за развој на дијабетес, вториот мора да се лекува, секогаш одржувајќи ја стапката на шеќер под контрола.
Ако се откриени патологии во црниот дроб, нејзиниот третман е во тек. Ова е неопходно со цел да се спречи развој на патологија до состојба на малиген тумор.
Хемохроматоза
Наследната хемохроматоза (НГ) е полисистемска болест базирана на генетски утврдени метаболички нарушувања на железо, што доведува до нејзино прекумерно акумулирање во организмот и токсично оштетување на органи и ткива.
Првиот опис на болеста му припаѓа на A. Trousseau (1865), кој идентификуваше тријада на главните клинички манифестации: дијабетес мелитус, бронзена пигментација на кожата, цироза. Терминот "хемохроматоза" беше предложен во 1889 година од Ф.Д. фон Реклингхаузен. Од 1935 година, болеста припаѓа на групата наследни заболувања. Во 1996 година, Ј.Н. Федер и сор. го идентификуваа генот за наследна хемохроматоза (HFE), чии мутации најчесто доведуваат до развој на оваа болест. Во 2000-2004 година опишани се мутации на други гени што доведуваат до развој на хемохроматоза.
Преваленцијата на болеста варира од 1: 250 лица кои живеат во Северна Европа до 1: 3300 кај црното население во САД и африканските земји. Болеста се дијагностицира кај мажите 5-10 пати почесто отколку кај жените. За време на генетското скринирање, откриено е дека хомозиготна мутација на генот HFE е откриена кај 1 од 500 испитани пациенти, додека бројот на клинички утврдени случаи на НГ е 1: 5000. Така, значителен број случаи на заболување не се препознаваат или дијагностицираат доцна, во фаза на неповратно внатрешно оштетување. органи (цироза, дијабетес мелитус, проширена кардиомиопатија).
Во согласност со генетската основа на болеста, се разликуваат 4 типа на наследна хемохроматоза:
Тип I - наследен од автозомно рецесивно механизам, заради мутации во генот HFE лоциран на хромозомот 6. Најчесто (кај 87-90% од пациентите) се запишува мутација на C282Y - замена на цистеин со тирозин во 282-та аминокиселина. Мутацијата H63D е поретка - замена на цитидин со гванин во 63-та аминокиселина,
Тип II - малолетничка хемохроматоза е ретка, поради мутации во генот одговорен за синтеза на друг протеин на метаболизам на железо - хепсидин,
Тип III - генетската основа се состои од мутации на синтеза на трансферин рецептор за кодирање на ген,
Тип IV - генетската основа се состои од мутации во генот SLC40A1, кој ја кодира синтезата на транспортниот протеин феропортин.
Етиологија и патогенеза
Ironелезото е неопходна биохемиска компонента од најважните метаболички процеси, од една страна, и е потенцијално токсичен елемент што може да предизвика оксидативно оштетување на биолошките мембрани, протеини и нуклеински киселини, од друга страна. Во согласност со ова, железната хомеостаза во човечкото тело е цврсто регулирана. Повеќето од овој елемент се подложува на процес на рециклирање: макрофаги на слезината и зафаќање на црниот дроб и уништува стари црвени крвни зрнца, го деградира хемоглобинот и ослободува железо, што се врзува за трансферин или феритин и се рециклира. Дневната физиолошка загуба на железо не надминува 1-2 мг и се компензира со апсорпција на еднаква количина железо во гастроинтестиналниот тракт. Не постојат механизми кои ја контролираат елиминацијата на железо кај луѓето.
Мутации на гените одговорни за синтеза на протеини кои се вклучени во метаболизмот на железо, доведуваат до нерамнотежа помеѓу внесот и губење на железо, патолошка акумулација на овој елемент во органи и ткива и појава на бесплатно (не поврзано со трансферин) железо во крвта. Развојот на хемохроматозата од типот I е поврзан со мутација на генот надлежен за синтеза на протеинот HFE (протеин на хемохроматоза), кој е гликопротеин (ММ = 37.235 далтони), сличен по структура со протеините на главниот комплекс за хистокомпатибилност од класа 1. Функцијата на протеинот HFE во метаболизмот на железо и механизмот на нагло зголемување на апсорпцијата на железо за време на мутациите во генот HFE не се целосно утврдени.
Патогенезата на хемохроматоза од типот II-IV е поврзана со мутации во гените кои кодираат други протеини вклучени во метаболизмот на железо - хепсидин, рецептор-трансферин-II, феропортин.
Карактеристична карактеристика на типот IV NG, која се заснова на мутации на генот на феропортин, е доминантно кршење на процесите за рециклирање на железо, што фенотипски се манифестира како длабока хипохромна анемија и еритропоеза со недостаток на железо во комбинација со тешка хемохроматоза на внатрешните органи.
Патолошката акумулација на железо во паренхимните органи е поврзана со дегенеративни промени во паренхимот на клетките и прогресивен развој на фиброзно ткиво, што доведува до неповратна дисфункција на виталните органи. Најранливи целни органи се црниот дроб, срцето и панкреасот.
Клинички знаци и симптоми
Клиничката слика на НГ се одредува според нивото на акумулација на железо во органи и ткива. Со хипертензија од типот I, клиничките манифестации обично се наоѓаат на возраст од 45-50 години и постари. Кај малолетничка хемохроматоза (тип II), сериозни лезии на црниот дроб и срцето се појавуваат рано - во втората или третата деценија од животот. Кај мажите, клиничките манифестации на болеста се забележуваат 3 пати почесто отколку кај жените, што е поврзано со физиолошките карактеристики на женското тело. Главните клинички манифестации вклучуваат симптоми на оштетување на црниот дроб, срцето, органи на ендокриниот систем и зглобовите.
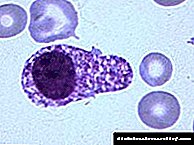 Знаци на оштетување на црниот дроб може да се открие за време на случаен преглед во форма на немотивирано зголемување на трансаминази или деби со симптоми на портална хипертензија: асцити, хепатоспленомегалија, крварење од проширени вени на хранопроводот и желудникот.
Знаци на оштетување на црниот дроб може да се открие за време на случаен преглед во форма на немотивирано зголемување на трансаминази или деби со симптоми на портална хипертензија: асцити, хепатоспленомегалија, крварење од проширени вени на хранопроводот и желудникот.
Симптоми на срцево оштетување вклучуваат срцев удар, развој на аритмии и знаци на срцева слабост. Тешката кардиомиопатија е водечка причина за смрт кај млади пациенти.
Развојот на дијабетес и дисфункција на гениталните жлезди се карактеристични симптоми на НГ. Кај мажите често се забележува атрофија на тестисите, намален сексуален нагон, импотенција, азоспермија, кај жени - аменореа, неплодност.
Оштетувањето на зглобовите се манифестира со постојана артралгија, најчесто се вклучени метакарпофалангеалните зглобови, поретко на коленото, колкот, лактот. Вкочанетоста на зглобовите постепено се развива.
Другите клинички манифестации на НГ вклучуваат изразена немотивирана слабост, замор, поспаност, периоди на болки во стомакот со различен интензитет и локализација, хиперпигментација на кожата и тенденција на разни инфекции (вклучително и микроорганизми кои ретко влијаат на здравите луѓе - Yersenia enterocolitica и Vibrio vulnificus).
Дијагнозата на НГ се утврдува врз основа на карактеристична клиничка и лабораториска слика.Лесно е да се сомневате во дијагноза на хемохроматоза кај пациент со комбинација на следниве симптоми: артралгија, болки во стомакот, бронзено-сива кожа, присуство на дијабетес мелитус и хепатомегалија.
Тест на крвта: карактеристична е комбинација на високо ниво на хемоглобин со ниска концентрација на хемоглобин во еритроцити (MCH). Развојот на анемија или друга цитопенија е забележан во доцните фази на болеста - кај пациенти со цироза на црниот дроб или е резултат на бројни крвавења.
Студија на метаболизмот на железо неопходно да се идентификуваат лабораториски знаци на преоптоварување со железо и вклучува определување на нивото на железо, феритин и трансферин на крвниот серум, вкупниот капацитет за врзување на железо на серум (OZHSS) и проценетиот коефициент на заситеност на трансферин на железо (NTZH) НГ се карактеризира со зголемување на нивото на серумско железо и феритин, намалување на нивото на OGSS и трансферин. Важен лабораториски знак на хемохроматоза е зголемување на коефициентот на СПИ кај мажи над 60%, кај жени - над 50%.
Десферален тест потврдува присуство на преоптоварување со железо: по интрамускулна 0,5 g дефероксамин (десферална), дневната екскреција на железо во урината значително го надминува нормалното ниво (0-5 mmol / ден).
Во типот IV NG, лабораториската слика може да биде претставена со длабока хипохромна анемија, хипосидеринемија и покачен серумски феритин, што е комбинирано со сериозно преоптоварување на ткивата со железо.
Спроведување молекуларна генетска анализа ви овозможува да ја потврдите наследната природа на хемохроматозата и да ја исклучите секундарната природа на преоптоварување со железо. Дијагнозата на NG е утврдена во присуство на хомозиготни мутации на генот HFE (C282Y или H63D) или кога сложени хетерозиготни (комбинација на хетерозиготни мутации C282Y и H63D) се откриени кај пациенти со лабораториски знаци на преоптоварување со железо. Изолирани хетерозиготни мутации C282Y и H63D се наоѓаат кај популацијата на здрави луѓе со фреквенција од 10,6% и 23,4% од случаите, соодветно, присуството на овие мутации не е основа за дијагностицирање на НГ.
КТ скенирање на абдоминални органи открива зголемена густина на ткивото на црниот дроб поради наслаги на железо и дозволува сомневање во присуството на хемохроматоза.
Со МНР црниот дроб на пациент со хемохроматоза има темно сива или црна боја. КТ и МНР на црниот дроб се неопходни за да се исклучи дијагнозата на хепатоцелуларен карцином.
Биопсија на црниот дроб со полу-квантитативно или квантитативно определување на содржината на железо ви овозможува да го одредите степенот на развој на фиброза и концентрацијата на железо во ткивото на црниот дроб. За дијагностицирање на хемохроматоза, се препорачува да се пресмета „индексот на хепатална железо“, што е еднакво на односот на содржината на железо во ткивото на црниот дроб (во сува тежина на микромол / g) со возраста на пациентот (во години). Индекс> 2.0 ја потврдува дијагнозата на НГ.
Наследната хемохроматоза мора да се разликува со сендроми на секундарно преоптоварување со железо, кои се развиваат кај пациенти со наследна и стекната хемолитичка анемија, некои форми на миелодипластичен синдром (огноотпорна сидеробластична анемија), порфирија, како и кај пациенти со алкохолно оштетување на црниот дроб.
Целта на третманот на НГ е да се отстрани вишокот железо од телото и да се спречи неповратно оштетување на внатрешните органи. Чест метод на лекување е крварење. Првичниот курс се состои од крварење во волумен од 500 ml еднаш неделно. По спуштање на нивото на хемоглобин за 15-20 g / l, нивото на MCV за 3-5 fl. и содржината на серумскиот феритин до 20-50 ng / ml, одете на терапија за одржување - отстранување на 500 ml крв на секои 2-4 месеци кај мажи и на секои 3-6 месеци кај жени. Третманот е доживотно.
Во присуство на анемија или други контраиндикации (на пример, срцева слабост), железни хелатори се користат за крварење. Дефероксамин врзува вишок железо во ткивата и крвниот серум и излачува со урина и измет. Сепак, полуживотот на оваа дрога е краток - само 10 минути, што бара бавна администрација: интравенски во форма на 3-4 часа инфузии или субкутано, по можност во форма на 12-часовни или деноноќни инфузии со употреба на специјални пумпи. Развиени се нови комплексни лекови за орална администрација и се во фаза на клиничка студија или примена, од кои најефикасен е Дефесаирокс.
Ефективноста на третманот е одредена од динамиката на клиничките и лабораториските податоци. Состојбата на пациентите започнува да се подобрува по текот на крварењето: слабоста, замор, поспаност исчезнуваат, големината на црниот дроб се намалува, може да се подобри текот на дијабетесот и кардиомиопатија. Лабораториска контрола вклучува проучување на хемограм, индикатори на феритин, железо и NTZH (1 пат за 3 месеци), нивото на екскреција на уринарно железо.
Во случај на рано дијагностицирање на хипертензија и навремено терапевтско крварење, прогнозата е поволна: животниот век на пациентите не се разликува од животниот век на луѓето кои не страдаат од хемохроматоза. Во случаи на доцна дијагностицирање на болеста, во присуство на цироза на црниот дроб, кардиомиопатија, дијабетес мелитус, прогнозата се утврдува со сериозноста на овие неповратни компликации. Главните причини за смрт на пациентите се: компликации на дијабетес, срцева слабост, примарен карцином на црниот дроб, слабост на црниот дроб, крварење од проширени вени на хранопроводот и желудникот, интеркурентни инфекции.
Општи информации
Хемохроматозата (бронзен дијабетес, пигментна цироза) е генетски предизвикано нарушување на метаболизмот на железо, што доведува до таложење на пигменти што содржат железо во ткивата и органите и развој на повеќекратна слабост на органите. Болеста, придружена со карактеристичен комплекс на симптоми (пигментација на кожата, цироза на црниот дроб и дијабетес мелитус) е опишана во 1871 година, а во 1889 година била наречена хемохроматоза за карактеристична боја на кожата и внатрешните органи. Фреквенцијата на наследна хемохроматоза кај популацијата е 1,5-3 случаи на 1000 популација. Мажите страдаат од хемохроматоза 2-3 пати почесто од жените. Просечната возраст на развој на патологија е 40-60 години. Поради полисистемската природа на лезијата, разни клинички дисциплини се вклучени во студијата на хемохроматоза: гастроентерологија, кардиологија, ендокринологија, ревматологија и др.
Во етиолошки аспект, се разликуваат примарна (наследна) и секундарна хемохроматоза. Примарната хемохроматоза е поврзана со дефект на ензимските системи, што доведува до таложење на железо во внатрешните органи. Во зависност од генскиот дефект и клиничката слика, се разликуваат 4 форми на наследна хемохроматоза:
- Јас - класичен автосомно рецесивен, HFE поврзан тип (повеќе од 95% од случаите)
- II - тинејџерски тип
- III - наследен HFE-неиздаден тип (мутации во рецепторот на трансферин тип 2)
- IV– автозомно доминантен тип.
Секундарната хемохроматоза (генерализирана хемозидероза) се развива како резултат на стекната инсуфициенција на ензимски системи вклучени во метаболизмот на железо, и честопати е поврзана со други болести, во врска со кои се разликуваат нејзините следни варијанти: пост-трансфузија, хранлива, метаболичка, мешана и неонатална.
Во клиничкиот тек, хемохроматозата минува низ 3 фази: I - без преоптоварување со железо, II - со преоптоварување со железо, но без клинички симптоми, III - со развој на клинички манифестации.

Причини за хемохроматоза
Примарна наследна хемохроматоза е автозомно рецесивно нарушување на преносот. Се заснова на мутации на генот HFE сместен на кратката рака на шестиот хромозом. Дефект во генот HFE доведува до нарушување на внесувањето железо со посредство на трансферин од страна на клетките на дуоденумот 12, што резултира во формирање на лажен сигнал за недостаток на железо во организмот. За возврат, ова придонесува за зголемена синтеза на протеинот што ја врзува железо ДЦТ-1 со ентероцити и засилена апсорпција на железо во цревата (со нормален внес на елементи во трагови од храна). Во иднина, постои претерано таложење на пигментот што содржи железо во хемосидерин во многу внатрешни органи, смрт на нивните функционално активни елементи со развој на склеротични процеси. Со хемохроматоза, 0,5-1,0 g железо се акумулира годишно во човечкото тело, а манифестациите на болеста се манифестираат кога ќе се достигне вкупното ниво на железо од 20 g (понекогаш 40-50 g или повеќе).
Секундарната хемохроматоза се развива како резултат на прекумерно егзогено внесување железо во организмот. Оваа состојба може да се појави со чести повторени трансфузии на крв, неконтролирано внесување на препарати од железо, таласемија, некои видови анемија, порфирија на кожата, алкохолна цироза на црниот дроб, хроничен вирусен хепатитис Б и Ц, малигни неоплазми, по диета со малку протеини.
Симптоми на хемохроматоза
Клиничката манифестација на наследна хемохроматоза се јавува во зрелоста, кога вкупната содржина на железо во организмот достигнува критични вредности (20-40 g). Во зависност од преовладувачките синдроми, се разликуваат хепатопатични (хемохроматоза на црниот дроб), кардиопатска (срцева хемохроматоза), ендокринолошки форми на болеста.
Болеста се развива постепено, во почетната фаза неспецифични поплаки преовладуваат за зголемен замор, слабост, слабеење, намалено либидо. Во оваа фаза, пациентите може да бидат вознемирени од болка во десниот хипохондриум, сува кожа, артралгија поради хондрокалциноза на големи зглобови. Во проширената фаза на хемохроматоза се формира класичен комплекс на симптоми, претставен со пигментација на кожата (бронзена кожа), цироза, дијабетес мелитус, кардиомиопатија, хипогонадизам.
Обично, најраниот знак на хемохроматоза е појава на специфична боја на кожата и мукозните мембрани, изразена главно на лицето, вратот, горните екстремитети, во пазувите и надворешните гениталии и лузни на кожата. Интензитетот на пигментацијата зависи од времетраењето на текот на болеста и варира од бледо сива боја (чад) до бронзено-кафеава боја. Карактеристично е губење на косата на главата и трупот, конкавна (лажица) деформација на ноктите. Забележани се артропатиите на метакарпофалангеалниот, понекогаш коленото, колкот и лактот и со последователен развој на нивната вкочанетост.
Скоро кај сите пациенти се открива зголемување на црниот дроб, спленомегалија, цироза на црниот дроб. Функција на панкреасот е изразена во развој на инсулин-зависен дијабетес мелитус. Како резултат на оштетување на хипофизата за време на хемохроматозата, сексуалната функција страда: кај мажите се развива атрофија на тестисите, импотенција, гинекомастија, кај жени - аменореа и неплодност. Срцевата хемохроматоза се карактеризира со кардиомиопатија и нејзините компликации - аритмија, хронична срцева слабост, миокарден инфаркт.
Во крајната фаза на хемохроматоза се развива портална хипертензија, асцити, кахексија. Смртта на пациентите, како по правило, се јавува како резултат на крварење од проширени вени на хранопроводот, откажување на црниот дроб, акутна срцева слабост, дијабетична кома, асептичен перитонитис, сепса. Хемохроматозата значително го зголемува ризикот од развој на карцином на црниот дроб (хепатоцелуларен карцином).
Дијагноза на хемохроматоза
Во зависност од преовладувачките симптоми, пациентите со хемохроматоза можат да побараат помош од разни специјалисти: гастроентеролог, кардиолог, ендокринолог, гинеколог, уролог, ревматолог и дерматолог. Во меѓувреме, дијагнозата на болеста е иста за различни клинички варијанти на хемохроматоза. По проценката на клиничките знаци, на пациентите им се доделува збир на лабораториски и инструментални студии за да се провери валидноста на дијагнозата.
Лабораториски критериуми за хемохроматоза се значително зголемување на нивото на железо, феритин и трансферин во крвниот серум, зголемување на излачувањето на железо во урината и намалување на вкупната способност за врзување железо на крвниот серум. Дијагнозата се потврдува со биопсија на пункција на црниот дроб или кожата, во примероците од кои е откриена таложење на хемозидерин. Наследната природа на хемохроматозата е утврдена како резултат на молекуларна генетска дијагностика.
Со цел да се процени сериозноста на оштетување на внатрешните органи и прогнозата на болеста, се изучуваат тестови на црниот дроб, нивото на гликоза во крвта и урината, гликозилиран хемоглобин, итн. Лабораториската дијагностика на хемохроматозата е дополнета со инструментални студии: зглобна радиографија, ЕКГ, ехокардиографија, ултразвук на абдоминалната празнина, црниот дроб МРИ, итн.
Третман на хемохроматоза
Главната цел на терапијата е да се отстрани вишокот железо од телото и да се спречи развој на компликации. Пациентите со хемохроматоза им е пропишана диета со која се ограничува храна богата со железо (јаболка, месо, црн дроб, леќата, спанаќ, итн.), Лесно сварливи јаглени хидрати. Забрането е земање мултивитамини, аскорбинска киселина, додатоци во исхраната кои содржат железо, алкохол. За да се отстрани вишокот железо од телото, тие прибегнуваат кон крварење под контрола на хемоглобин, хематокрит и феритин. За таа цел, може да се користат екстракорпорална методи на хеморекција - плазмафереза, хемосорпција, цитафереза.
Патогенетска терапија со лекови на хемохроматоза се заснова на интрамускулна или интравенска администрација на дефероксамин врзувачки јони на дефероксамин на пациент. Во исто време, се спроведува симптоматски третман на цироза на црниот дроб, срцева слабост, дијабетес мелитус и хипогонадизам. Со тешка артропатија се утврдуваат индикации за артропластика (ендопростетика на зафатените зглобови). Кај пациенти со цироза, се третира прашањето за трансплантација на црниот дроб.
Предвидување и спречување на хемохроматоза
И покрај прогресивниот тек на болеста, навремената терапија може да го продолжи животот на пациентите со хемохроматоза за неколку децении. Во отсуство на третман, просечниот животен век на пациентите по дијагностицирање на патологија не надминува 4-5 години. Присуството на компликации на хемохроматоза (главно цироза на црниот дроб и конгестивна срцева слабост) е прогностички неповолен знак.
Со наследна хемохроматоза, превенцијата се сведува на скрининг во семејството, рано откривање и лекување на болеста. Рационална исхрана, следење на администрацијата и администрацијата на препарати од железо, трансфузија на крв, одбивање на алкохол и следење на пациенти со заболувања на црниот дроб и крвниот систем може да помогнат во спречување на развој на секундарна хемохроматоза.


-
Како брзо да го отстраните холестеролот од телото
Како да го отстраните холестеролот од телото За да го отстраните холестеролот од телото, треба да ја намалите телесната тежина, да ја зголемите физичката активност и да се ослободите од лошите навики. ... -

-

-